
Regulatory Agency's official website link:https://www.fda.gov/drugs/forms-submission-requirements/drug-master-files-dmfs
Database link: https://www.fda.gov/drugs/drug-master-files-dmfs/list-of-drug-master-files-dmfs
English.
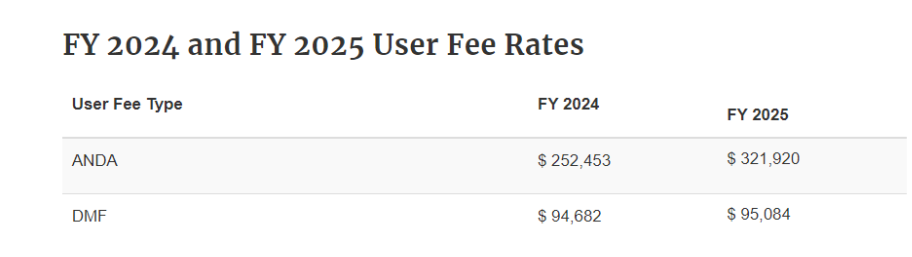
The user fees for Type II API DMF reviews vary by U.S. fiscal year (October 1 to September 30 of the following year).
In terms of review timelines, the administrative review is generally completed within 2–3 weeks after submission. For the Completeness Assessment (CA) review, API Type II DMFs require payment of GDUFA fees before the CA review can proceed, which typically takes about 60 days.
Q: At the time of applying for a 510K, must the technical parameters of the device submitted for application be exactly the same as the technical parameters of the consistent device?
A: The FDA allows some variation in technical parameters, but it must be demonstrated that these differences do not raise safety and efficacy concerns with the comparison device.
Q: How to deal with that, after submitting the 510K application, FDA determines that the device is substantially not equivalent?
A: Additional information or additional testing: After a challenge from the FDA, the manufacturer is required to provide additional information or perform additional testing to demonstrate the safety and efficacy of the device. This may include providing additional clinical data, laboratory test results or other relevant evidence 12.
Modification of design or manufacturing process: If FDA determines that a device has significant differences in technical characteristics from a consistent device, the manufacturer may need to modify the design or manufacturing process to ensure that the device is consistent with the generic device in technical characteristics 1.
Resubmit the application: After adding the necessary information or performing additional tests, the manufacturer may resubmit the application. Ensure that all documentation is complete, accurate, and clearly shows the device's similarity to consistent devices 23
Q: I want to distribute a product for which another manufacturer has obtained 510K approval and reflect my company's name on the labeling, does it require to reapply a 510K?
A: There are no significant changes to the labeling (e.g., intended use, indications, population, contraindications, contraindications, etc.) and "Distributed by ABC Firm" or "Manufactured for ABC Firm" (21 CFR 801.1) on the labeling, a new 510K application is not required.
Q: What is a U.S. agent and what are his duties?
A: U.S. Agent means a foreign manufacturer | brand | trader who has business in or in the United States is appointed as a registered agent for the purpose of FDA registration. U.S. Agent is a prerequisite for FDA registration.
U.S. agents act as the communication link between FDA and foreign manufacturers | brands | traders. They are not only required to submit application information at the time of registration, but are also responsible for emergency and routine communication. Their duties are as follows:
Document preparation and submission: Responsible for submitting documents required for product registration to FDA. Such as 510 (k) Advance market notification, PMA (Pre-Market Approval) application, alert notification, recall notification and other relevant documents.
Document maintenance and records: Maintain records of these documents for FDA audit and inspection to ensure that documents are available when required.
Information transfer and communication: Responsible for receiving and conveying all information, requirements and notices from FDA to manufacturers, submitting all necessary documents, notices and applications to FDA on behalf of manufacturers, and handling all correspondence and inquiries with FDA, including registration, license, application and other aspects of communication.
Problem solving assistance: Assist medical device manufacturers in resolving and responding to problems with the FDA in the event of product quality, safety, or regulatory issues. Documentation and information may be required to help FDA understand the situation and facilitate resolution of the issue.
[Note] : The FDA will contact the U.S. agent from time to time, and the U.S. agent must be able to answer the phone call from the FDA at any time. For the agent with untrue information, the FDA will require the factory to provide true information, otherwise it will impose penalties or even cancel the registration number of the factory.











































