Indonesia Medical Device Regulatory Authority
The primary government agency responsible for medical device regulation in Indonesia is the Ministry of Health of the Republic of Indonesia (MOH). This agency is responsible for pre-market and post-market assessment, standardization, legislation, and Good Manufacturing Practice (GMP) certification for medical devices.
According to Article 5 of the Ministry of Health Regulation No. 5 of 2022, the Indonesian MOH performs the following functions:
① Formulating, determining, and implementing policies in the areas of public health, disease prevention and control, health services, pharmaceuticals, medical devices, and health personnel, coordinating the execution of policy tasks, providing guidance and administrative support to all organizational units within the Ministry;
② Managing state property under the responsibility of the MOH;
③ Overseeing the implementation of the MOH's duties;
④ Providing technical guidance and supervision on the execution of MOH affairs at the regional level;
⑤ Formulating and providing recommendations on health development policies;
⑥ Providing substantive support to all units within the MOH.
Any medical device, in vitro diagnostic (IVD) device, and PKRT (Household Health Supply/Perbekalan Kesehatan Rumah Tangga) product entering the territory of the Republic of Indonesia must be registered and obtain a distribution permit (marketing authorization) before being sold in the Indonesian market.
Regulatory Framework
Regulation of the Minister of Health No. 62 of 2017 concerning Medical Devices and Household Health Supplies
This regulation stipulates requirements for the registration, classification, production, distribution, and supervision of medical devices, covering definitions, classification criteria, registration procedures, and market access requirements. Indonesia also references relevant ASEAN regulations (such as the ASEAN Medical Device Directive), as well as specific registration and supervision guidelines.
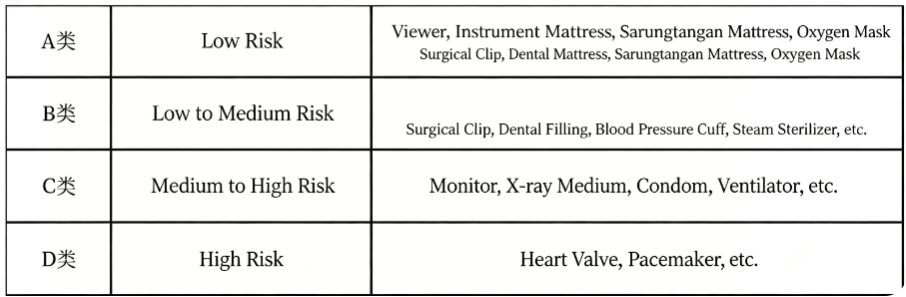
Risk Classification of Medical Devices and IVD Products
Based on the potential risk posed to patients by the use of medical devices, the classification from low to high is as follows:
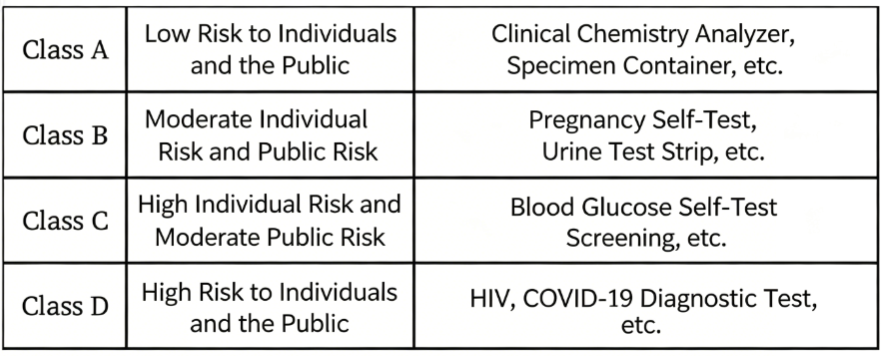
Based on the risk of misinterpretation of examination results for individuals and the public, in vitro diagnostic (IVD) medical devices are classified as follows:
Registration Basis
According to Indonesian medical device registration regulations, product registration in the country of origin is not a prerequisite for submitting an application in Indonesia. However, regardless of the product's risk classification, a notarized Free Sales Certificate (FSC) must be provided when submitting the application. Regarding the issuing authority of the FSC, in principle, a document issued by the regulatory authority of the country of origin is required. If the product is not registered or marketed in the country of origin, but has been registered in any one of the following countries/regions – the European Union, the United States, Australia, Canada, or Japan – the corresponding FSC issued by the regulatory authority of that country may be submitted as an alternative.
Admission Prerequisites
When selling medical devices in Indonesia, a foreign manufacturer must appoint a sole local agent. This agent will assume local responsibility for product registration, distribution, and after-sales service.
Establish a quality management system compliant with ISO 13485 to ensure product quality.
Prepare detailed technical documentation based on the ASEAN Common Submission Dossier Template (CSDT), and in compliance with Indonesian regulations and standards, including product design, specifications, manufacturing processes, material composition, performance characteristics, clinical trial data (if applicable), etc.
For certain high-risk or specific medical devices, clinical trial data may be required to demonstrate product safety and efficacy. Clinical trials must be conducted by approved institutions and comply with ethical and regulatory requirements.
Language Requirements for Registration
English + Indonesian
Indonesia Sole Agent/Sole Distributor/Exclusive Distributor
A Sole Agent/Sole Distributor/Exclusive Distributor refers to a PAK or PKRT importer appointed by a producer, manufacturer, or principal, acting as a representative to register and distribute medical devices, in vitro diagnostic medical devices, and PKRT within the territory of the Republic of Indonesia, and to provide after-sales services for medical devices, in vitro diagnostic medical devices, and PKRT.
If the product license application is submitted by a PAK or PKRT company appointed as a Sole Agent/Sole Distributor/Exclusive Distributor and/or authorized to register, the validity period of the product license shall be the same as the validity period of such appointment or authorization.
The product license is declared invalid under the following circumstances:
① The product license expires;
② The manufacturing certificate expires;
③ The PAK license expires;
④ The appointment as General Agent/General Distributor/Exclusive Distributor and/or authorization expires or is not renewed; or
⑤ The product license is revoked.
Registration Process, Timeline and Official Fees
1. Registration Process
Pre-registration Stage: Pre-registration is the process whereby the evaluator verifies the risk classification of the medical device to determine the Non-Tax State Revenue (PNBP) fee.
① The applicant shall submit an application through the website https://regalkes.kemkes.go.id/ as required. The pre-registration evaluation result will be notified via the website and email. The applicant should actively check and verify the evaluation result.
② The evaluator shall classify the risk of the medical device within a maximum of 7 days.
③ The applicant will receive an email notification of the Non-Tax State Revenue fee, which must be paid according to the classification of the medical device.
④ The applicant must pay the Non-Tax State Revenue fee and upload the payment receipt within 14 days of receiving the fee notification.
⑤ During the pre-registration stage, the evaluation and verification of the completeness of the submitted data has not yet begun.
For registration dossiers that do not meet the requirements, two opportunities to submit supplementary data will be given, and each supplementary submission must be completed within 30 days.
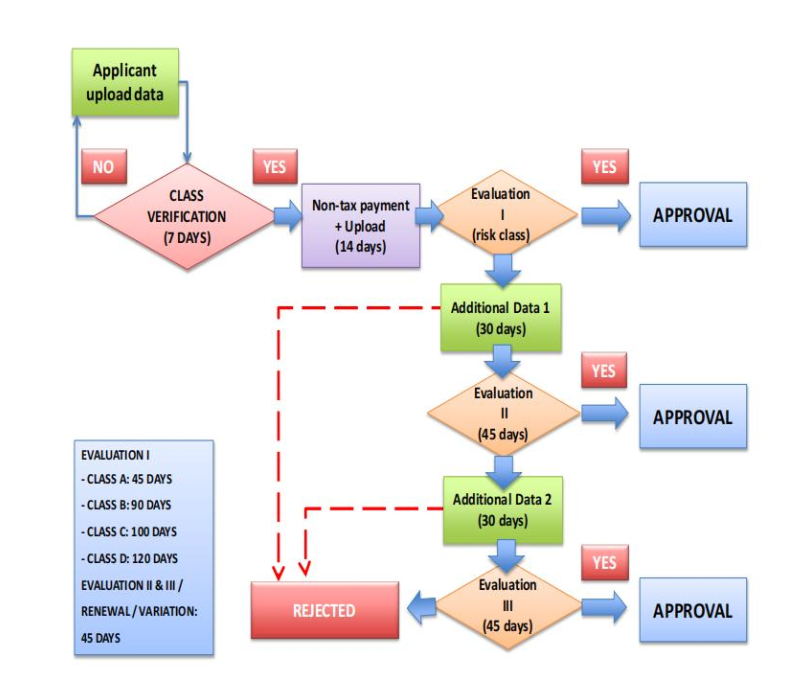
2. Registration Timeline
The requirements and length of time to obtain marketing authorization depend on the risk classification of the medical device.
3. Official Fees