What are IVD In Vitro Diagnostic Products?
IVD registration refers to the whole process of submitting in vitro diagnostic products to competent regulatory authorities (including FDA, CE, NMPA, etc.) and obtaining marketing licenses after official review, evaluation and certification to legally sell and distribute the products on the market. This process must comply with relevant laws, regulations and specifications, including ISO 13485 Quality Management System, ISO 14971 Risk Management System, Good Manufacturing Practice (GMP), etc.
The IVD registration workflow covers application submission, official review, technical evaluation and license issuance. Applicants shall submit complete product dossiers and supporting documents, including product technical specifications, safety assessment reports, clinical trial data, quality assurance plans and other materials. Regulators will conduct full review and technical assessment, with possible outcomes including full approval, supplementary data request or rejection. Once approved, the applicant will be granted a marketing license authorizing commercial sales and clinical use.
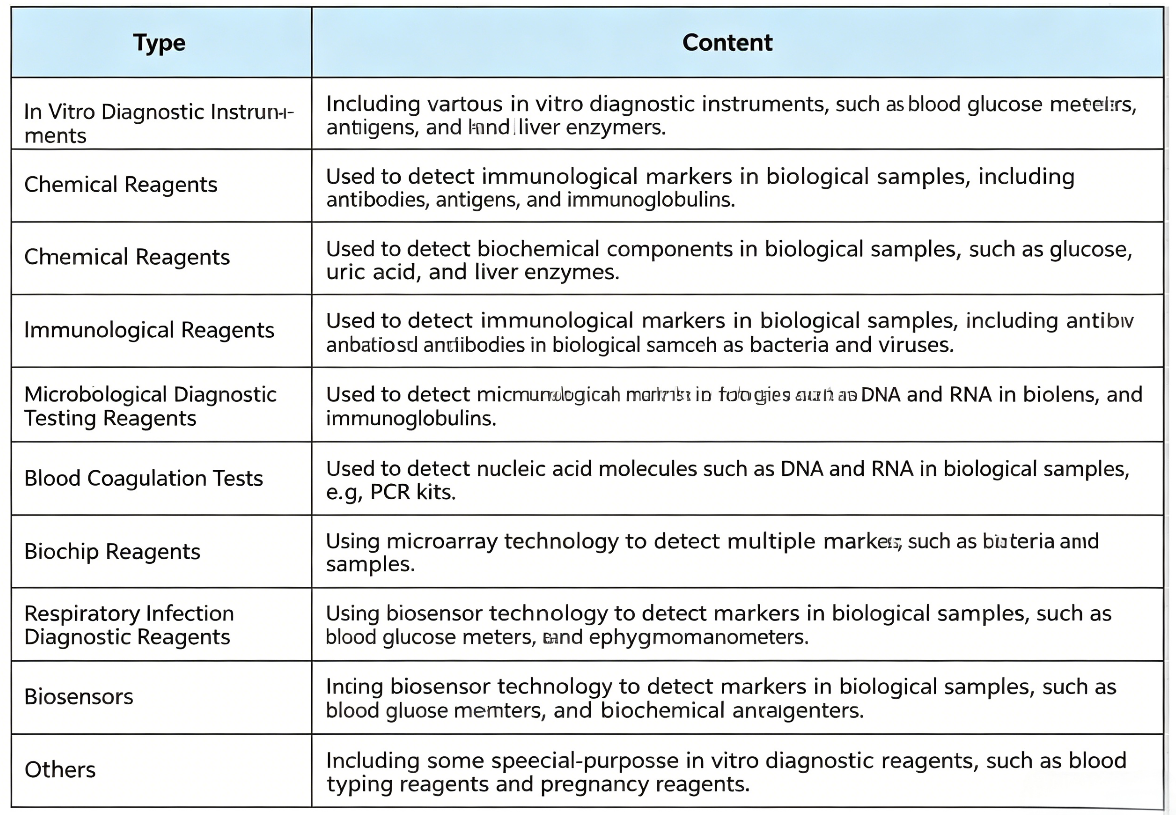
Classification standards for IVD products vary across countries, regions and industries. Based on product characteristics and intended use, IVD products are categorized as follows:
Detailed Procedures for Clinical Trials & Registration of IVD Products
- Develop Clinical Trial Protocol & Study Design When drafting the trial protocol and study design, developers shall clarify trial objectives, study subjects, sample size, research methodology and data analysis plans. All protocols must fully comply with ethical and legal requirements.
- Select a Suitable Clinical Trial Registry Multiple international IVD clinical trial registries are available, such as ClinicalTrials.gov and EU Clinical Trials Register. Registry selection shall take into account platform credibility, registration procedures and administrative fees.
- Register the Trial & Submit Required Documentation Applicants shall submit full documents including trial protocol, study design, research objectives and anticipated trial results, ensuring all information is accurate and complete. After registration, the full trial protocol will be publicly accessible and searchable on the registry platform.
- Obtain Ethics Committee Approval Prior to initiating any IVD clinical trial, ethics committee approval is mandatory. The committee will review the trial protocol and study design to verify full compliance with ethical and legal standards.
- Conduct the Clinical Trial During trial execution, researchers shall strictly follow the approved protocol and study design to collect, analyze and report all clinical data.
- Update Registry Information Timely Any revisions to the trial protocol or study design during implementation must be updated on the registry with revised documents submitted to the registry authority without delay.
- Complete the Trial & Submit Evaluation Data Upon trial completion, developers shall analyze and evaluate all clinical results and submit full evaluation reports to relevant authorities (regulatory bodies, academic institutions, medical organizations, etc.). Authorities will verify the scientific validity and reliability of trial data to safeguard public health and safety.
Documentation Required for International IVD Registration
Required registration dossiers differ by jurisdiction, but generally include the following materials:
- Product Registration Application Form: Complete basic product information including product name, model, intended use and target population.
- Instructions for Use (IFU): Detailed descriptions of product performance, operating procedures, warnings and precautions.
- Product Labelling & Packaging: Labels and packaging shall comply with local regional standards covering text, graphics, barcodes and other marking requirements.
- Product Test Reports: Full testing records covering performance indicators, safety and clinical effectiveness.
- Manufacturer Quality Management System Certificates: Valid certification documents such as ISO 13485 certificates.
- Manufacturer Business License & Production Permit: Official business registration and medical device production licenses issued by competent authorities.
- Local Marketing Authorization Certificate: Many jurisdictions require a regional sales license prior to commercial launch, with corresponding supporting documents to be provided.
- Other Supporting Documents: Jurisdiction-specific supplementary materials, such as CE marking certificates, FDA clearance certificates, etc.
International IVD clinical trial and registration projects require careful registry selection, consistent accuracy and completeness of submitted data, timely updates of trial information and full submission of post-trial evaluation reports. All work must adhere to ethical and legal regulations to guarantee the scientific integrity and reliability of clinical trial data.