
As a non-EU member state, Switzerland’s medical device regulatory system is independent of the EU yet closely aligned with international standards, giving it a significant standing in the global healthcare sector. Swissmedic is the central authority responsible for the registration and regulation of medical devices; it is renowned for its rigorous approval standards and transparent processes, and enjoys a particularly high international reputation in the fields of innovative devices and high-risk medical technologies. Although Switzerland has not adopted the EU Medical Device Regulation (MDR/IVDR), in practice it draws extensively on EU standards and requires non-Swiss manufacturers to appoint a local authorised representative (CH-REP) to assist with the registration process.
Regulatory authority website: https://www.swissmedic.ch/swissmedic/en/home.html
Database link: https://swissdamed.ch/
Medical Devices Ordinance (MedDO)
Ordinance on In Vitro Diagnostic Medical Devices (IvDO)
The classification of medical devices (including in vitro diagnostic devices) is governed by Article 15 of the Medical Devices Act (MedDO) and Article 14 of the In Vitro Diagnostic Medical Devices Act (IvDO). These provisions correspond to Annex VIII of the EU MDR and the EU IVDR respectively, and are broadly consistent with the EU classification rules.

English; the labelling must be in one of the official languages of Switzerland (German, French or Italian).
In Switzerland, manufacturers, importers or authorised representatives of medical devices may submit registration applications as applicants. If a medical device manufacturer does not have a place of business in Switzerland, its products may only be placed on the market once an authorised representative located in Switzerland has been designated (Article 51(1) of the Medical Devices Ordinance).
Registration Process
In accordance with the Swiss MedDO regulations and the phased requirements of the SwissDamed database, the medical device registration process comprises two procedures—a simplified procedure and a full procedure—depending on the product’s risk classification. The process can be broadly divided into five core steps, as detailed below:
1. Preliminary Preparation and Entity Registration: Non-Swiss manufacturers must first determine the product’s risk class and then appoint a Swiss Authorised Representative (CH-REP).
2. Preparation of Technical Documentation: Class I low-risk devices require only a basic declaration of conformity and product compliance documentation; Class IIa and above devices require a full set of technical documentation, including design documentation and ISO 13485 quality management system certification; Class III devices must additionally provide a clinical evaluation report demonstrating suitability for the Swiss population. In addition to English documents, labels and instructions for use must be provided in German, French, Italian or any one of these languages, and must include the CH-REP details.
3. Selecting the procedure and submitting the application: Holders of a CE certificate may follow the simplified procedure, submitting the CE certificate as the core documentation; those without a CE certificate must follow the national procedure, requiring the additional submission of a risk-benefit analysis report, etc. Applications are submitted via the Swissmedic e-portal, and fees must be paid according to the risk category (generally between 6,000 and 20,000 Swiss francs).
4. Review and Certificate Issuance: Class I devices do not require prior review by Swissmedic and can complete compliance registration within 2–4 weeks; the review process for Class IIa devices takes approximately 90 days, whilst Class III devices may take up to 120 days. Should issues arise during the review, the company must submit corrective documentation within the specified timeframe; upon successful review, Swissmedic will issue a registration certificate.
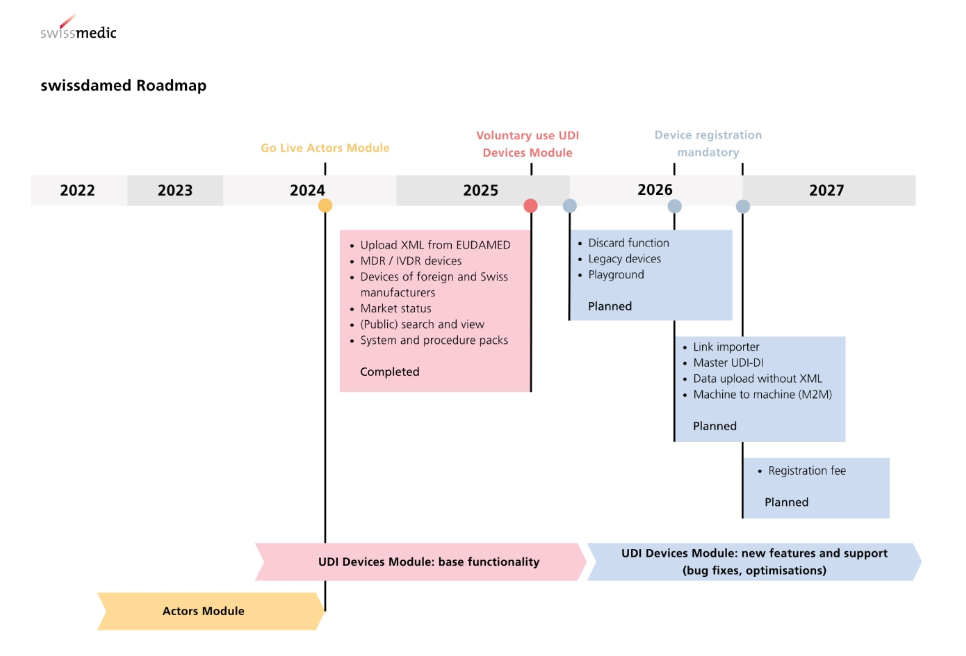
5. Device Registration and Post-market Surveillance: Currently, it is voluntary to register device information via the UDI module on Swissmedic’s platform; however, this will become mandatory from 1 July 2026. Following market authorisation, Class III devices must submit annual safety update reports, and all devices must report serious adverse events within 24 hours; significant product changes also require resubmission for review.
A: Yes.
A: If the medical device product changes meet the requirements, registration changes can be made according to the procedure.










































