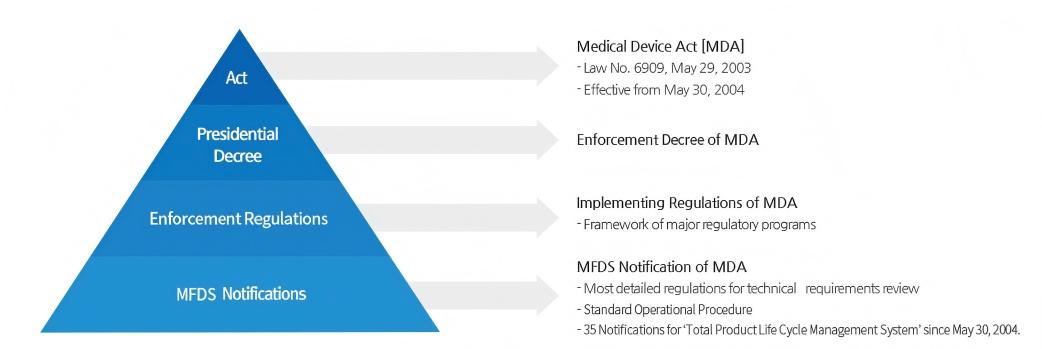
Enforcement Decree of the MDA
【Medical Device (except IVD)】

【IVD(In Vitro Diagnostic Medical Devices)】

Korean and English.



Official Review Time (Based on Wiselink Project Experience)
Medical devices in South Korea are classified by risk level, each corresponding to different market access pathways with varying review durations:
① Class I Devices: Approximately 2 months;
② Class II Devices: Approximately 4–7 months;
③ Class III and IV Devices: Approximately 145 business days.

A: The registration of medical devices in Korea is mainly approved by MFDS, but the audit bodies corresponding to products with different risk levels are different.
① Class I medical devices: Certified by the Medical Device Information and Technical Assistance Center (MDITAC), the audit time is approximately 2 months.
② Class II medical devices: Certified by MDITAC, the audit time is approximately 4-7 months. In addition, the certification method of Class II devices is similar to that of the US FDA 510K, which proves the safety and effectiveness of the product by comparing the consistent products.
③Class III and Class IV medical devices: Directly approved by MFDS, the audit time is approximately 145 workdays. Class III and Class IV devices are subject to safety and effectiveness audit.
A: A total of 52 types, including 48 types of medical device which implanted in the human body for more than one year and 4 types of life-supporting devices that can be used outside of medical facilities, for which the relevant parties are required to keep records in accordance with the regulations, including the number of products, the date, the user and other information.
A: Yes.
A: Korean medical device registration certificates are usually valid for 5 years. After the expiration, the manufacturer needs to apply for re-registration or renewal to maintain the market eligibility of the product. Renewal applications must be submitted before the end of the registration period to avoid registration lapses and disruption of marketing.
During the renewal process, the Korea Food and Drug Safety (MFDS) may request to update technical documentation、 test data, or other relevant information. Renewal applications need to be submitted in advance of the expiration of the certificate to ensure that renewal approval is obtained before the expiration of the validity period
A: The GTHF's rules do not directly speed up the audit process in Korea. However, understanding and following the GHTF rules will help companies better prepare registration materials and improve the quality of registration applications, which may indirectly improve registration efficiency. First, although Korea authority will refer to GHTF's classification and principles for medical device registration, the specific registration requirements and processes are still determined by Korean regulations. Second, in order to speed up the registration audit of medical devices, the fast-track or priority audit mechanism provided by the MFDS in Korea is usually based on the urgency, innovativeness, or importance of the product rather than whether the company understands the rules of the GHTF.
A: Yes. However, it requires the consent and authorization of the original holder, and it needs to be notarized
A: KGMP certification requires a manufacturer's on-site audit.
According to the Korean Medical Device Act, Class III and IV medical devices are subject to the MFDS approval process, while Class I and II devices are certified and approved by the Medical Device Information and Technical Assistance Center (MDITAC) and MFDS, respectively. Manufacturers must undergo an on-site audit by an MFDS accredited body to obtain a KGMP certificate.
Among them, manufacturers of Class II, III and IV medical devices are required to conduct quality system conformity assessment to ensure that their quality systems comply with KGMP standards. The corresponding manufacturer must undergo an on-site audit to verify that its production process and management system comply with the requirements of KGMP.
In addition, to streamline the audit process, the MFDS allows desktop audits using the results of the Medical Device Single Audit Program (MDSAP) audits, but this is only applicable to initial audits, additional audits, and change audits, and renewal audits must still be conducted on-site.










































