Saudi Arabian SFDA administration information abstract

Administrative Regulation
[Medical Devices Law]
[Implementing Regulation of Medical Devices Law]
Classification of Risk Levels for Medical Devices and IVD Products
Saudi Arabia divides medical device MD and in vitro diagnostic reagent IVD into categories A, B, C and D according to the risk level of medical devices from low to high.

According to the risk level of medical devices from low to high, in vitro diagnostic reagents IVD is divided into:

Entry requirements
Product list, design description, accessories, materials, drawings, packaging, photos, risk analysis and other various reports, as well as certificates, plans, test reports, clinical evaluations, checklists, statements, etc., cover many aspects ranging from product information to various certification and assessment requirements, to ensure that medical devices comply with relevant standards and regulations, and guarantee their safe and effective launch and use.
Saudi Arabia does not mandate the use of CE, but if the product label bears the CE mark, evidence must be provided to support it.
Registration Language
English; If the medical device is for home use or for use by non-professionals, Arabic translations of the labels, instructions for use, and marketing materials are required.
Saudi representative
An authorized representative (AR) refers to any natural person or legal entity established within Saudi Arabia (KSA), who has received written authorization from the manufacturer to perform specific tasks, including the obligation to communicate on behalf of the manufacturer with the SFDA.
Responsibilities:
① It is necessary to have an AR business license (MDEL) within the territory of Saudi Arabia. If the authorized representative acts on behalf of multiple manufacturers, separate licenses need to be applied for for each manufacturer. If there is any update to the MDEL, it is necessary to inform the SFDA. The validity period of the AR business license (MDEL) is one year. Applicants can apply for renewal within 60 days before the expiration of the AR business license.
② A written authorization is required, which specifies the designated responsibilities of the authorized representative. The establishment and compliance with the relevant procedures (SOP) for authorization activities need to meet the requirements of the quality management system.
③ The manufacturer can choose to appoint an authorized representative to represent all the medical devices, or to designate different authorized representatives for each device category.
Registration process, cycle and official fees
① Must have AR business license MDEL in Saudi Arabia, if authorized to represent multiple manufacturers, need to apply for a separate business license for different manufacturers. SFDA should be informed of any updates from MDEL. AR business license MDEL is valid for 1 year. Applicants may apply for renewal of AR business license within 60 days prior to expiration.
② A written authorization is required which sets out the designated duties of the authorized representative. Establish and comply with procedures and SOP for authorized activities to meet the requirements of the quality management system.
③ The manufacturer can choose to appoint one authorized representative to represent all medical devices, or to appoint a different authorized representative for each device class.
Registration Flow、Cycle & Official Fee
1. Registration Flow
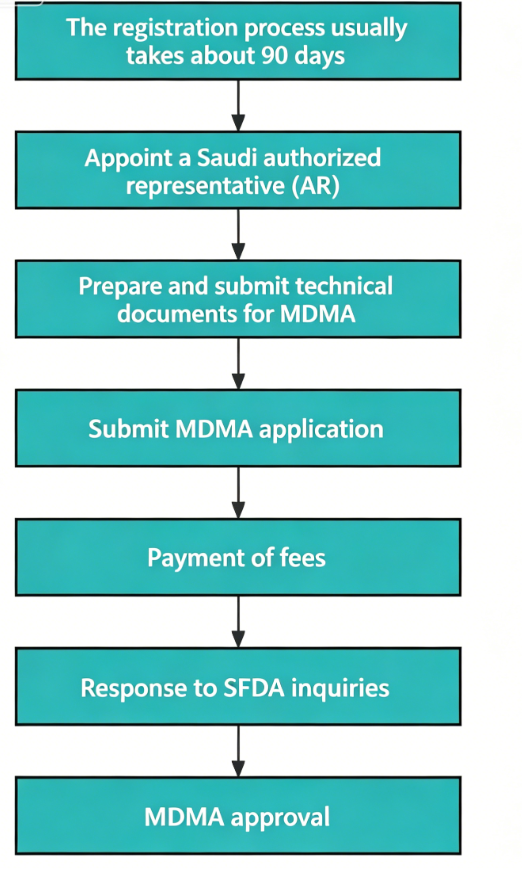
2. Cycle & Official Fee
Registration period: The approval time for MDMA is 2-3 months after submitting the application materials. The approval time for products with higher risk levels will be longer.
Official fees:
Annual fee for AR: 1840 USD
Registration assessment fee for Class A: SAR 1500
Registration assessment fee for Class B: SAR 1900
Registration assessment fee for Class C: SAR 2100
Registration assessment fee for Class D: SAR 2400
七、FAQ
Q: What is a License to Operate (LTO)? Is the customs clearance document (Declaration of conformity) the same as the registration document?
A: The customs clearance document is different from the "declaration that the goods comply with medical device and product control regulations", which refers to the registration and licensing information of the products in the goods, as well as the documentation of the manufacturer confirming that the products are shipped by the importer.
Q: What does a post-market evaluation study mean?
A: These are studies conducted on medical devices using scientific methods to verify their safety/effectiveness because of the presence of safety signals on the devices sold locally.
Q: What are the expected outcomes of the post-market evaluation study?
A: Corrective action;Preventive measure;Additional clinical studies are required to provide evidence of the safety and effectiveness of medical devices;Disseminate secure communications to users and healthcare providers.
Q: If a manufacturer sells a medical device directly to a buyer without an authorized representative (AR), who is responsible for notifying the affected buyer and providing a corrective action plan and closure?
A: Manufacturers are responsible for medical devices sold in Saudi Arabia, as well as suppliers and distributors.
Q: What procedures are followed for appointing a manufacturer liaison officer or his authorized representative to SFDA?
A: You must comply with the Medical Device Post Marketing Supervision Person Notice, sign the authorization agreement and submit it to SFDA to obtain the ARL (Authorized Representative License).
ARL require Apostille.










































