The registration of medical devices is primarily regulated by the UK’s MHRA, which employs a risk-based classification system (Class I, IIa, IIb and III). The route to market is determined according to the respective category; low-risk products may be subject to self-declaration, whilst medium- and high-risk products must be certified by a UK Approved Body and bear the UKCA mark. Manufacturers must appoint a UK Responsible Person to fulfil registration and compliance obligations, and submit applications via the MHRA’s online system. Following registration, they must regularly update information and undergo supervision, whilst complying with ongoing compliance requirements such as labelling, UDI and post-market surveillance. (Note: During the Brexit transition period, CE certificates are accepted for registration with the MHRA.)
MHRA: The Medicines and Healthcare products Regulatory Agency (MHRA) is responsible for operating the UK Medical Device Vigilance System, including conducting market surveillance, enforcing regulations, and collaborating with healthcare and regulatory stakeholders in the UK and globally.
Although the MHRA does not certify medical devices, it supervises UK Approved Bodies. If the MHRA considers a medical device to be unsafe, it has the authority to remove it from the UK market.
UKCA: UK Conformity Assessment is the abbreviation for UK Conformity Assessment. It is the UK product marking used for certain goods (including medical devices) sold on the UK market (England, Wales and Scotland).
Regulatory authority website link:https://www.gov.uk/guidance/register-medical-devices-to-place-on-the-market
Database link: https://pard.mhra.gov.uk/

All medical devices, including IVDs, custom-made devices, and systems or kits, must comply with the requirements of the Medical Devices Regulations 2002 (SI 2002 No 618, as amended) (UK MDR 2002) in order to be placed on the market and registered with the MHRA in the UK. This means that medical devices must obtain a UKCA or CE certificate before entering the UK market. This certificate demonstrates that the device complies with the relevant regulations.
English.
Manufacturers outside the UK must appoint a UK Responsible Person, i.e. a local authorised representative in the UK, to register their products.
① The UK Responsible Person must provide written evidence demonstrating that they have been authorised by the manufacturer to act as their UK Responsible Person. The UK Responsible Person carries out specific tasks on behalf of the non-UK manufacturer relating to the manufacturer’s obligations; this includes registering the manufacturer’s devices with the MHRA before the devices enter the UK market.
② Importers and distributors are not required to appoint a UK Responsible Person.
③ In addition to the registration requirements, the UK Responsible Person must:
a) Ensure that a Declaration of Conformity and technical documentation have been drawn up and, where applicable, that the manufacturer has carried out the appropriate conformity assessment procedures
b) Provide the MHRA, upon request, with all information and documentation necessary to demonstrate the conformity of the devices
c) cooperate with the MHRA in taking any preventive or corrective measures to eliminate, or where this is not possible, mitigate the risks posed by the device
d) immediately inform the manufacturer of any complaints and reports from healthcare professionals, patients and users regarding suspected incidents relating to the devices under their responsibility
① If the UK importer is not the UK authorised representative, the importer must inform the relevant manufacturer or UK authorised representative of their intention to import the device. In such cases, the manufacturer or the manufacturer’s UK authorised representative must provide the MHRA with the importer’s details. Further guidance on device registration.
② The obligations regarding the storage, transport and inspection of device labels bearing the CE mark or UKCA mark also apply. The importer’s or distributor’s name and address need not appear on the label.
Medical Device MHRA Regulatory Standard Terminology:
1.Confirm product classification and registration requirements per the classification rules adopted by MHRA, and retrieve the GMDN Code
2.Sign an agreement with the UK Responsible Person (UKRP)
3.Compile MHRA registration documentation
4.Submit registration documentation to MHRA
5.MHRA Review
Branch 1: Certificate Issued Upon Successful Review
Branch 2: MHRA issues feedback, requesting document revision or supplementary materials
Sub-branch A: Application rejected with supporting reasons
Sub-branch B: Certificate Issued Upon Successful Review
Registration period:
The entire registration process takes 1–2 months, depending on the complexity of the product.
Official fees:
Annual fee: £300 per GMDN sub-category per year (for registrations submitted during the financial year, the annual fee is calculated pro rata based on the number of days remaining)
1. From 1 April 2026: The fee structure will change, with the key criterion being whether the change introduces a new ‘GMDN sub-category’:
- New chargeable items: Primarily for adding devices falling under a new GMDN sub-category, or where a change results in the device requiring re-registration and falling under a new GMDN category.
- Now free of charge: Changes to address and company name will no longer incur a fee.
- Conditional exemption: For example, changes to regulatory status or IVD status; if the GMDN classification of the device after re-registration remains the same as before the change, no fee will be charged.
2. Items that are always free of charge: These include updating contact details, adding product models to registered devices, removing devices, updating obsolete GMDN terminology, linking new conformity assessment documents (where the regulatory status remains unchanged), and account deactivation. No statutory fees will be charged for these operations at any time.
Medical devices sold on the UK market must bear either the ‘UKCA’ mark or the ‘CE’ mark, depending on which regulation the device has been certified under. However, the ‘CE’ mark and the ‘UKCA’ mark may appear together on the device’s label.
Where applicable, the number of the UK Approved Body or Notified Body must also appear on the label.
The name and address of the UK Authorised Representative for devices bearing the UKCA mark must be included on the product label or outer packaging, or in the instructions for use. Where the CE mark is used, the details of the UK Authorised Representative need not be included at this stage.
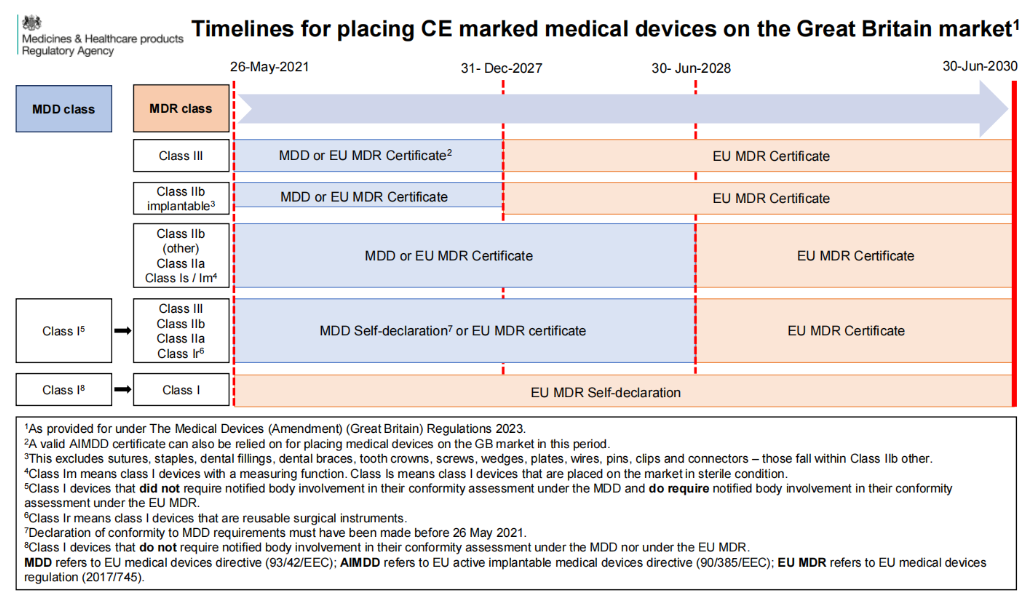
Transitional policy for devices that have obtained EU CE certification (including medical devices and IVDs)

A: For low-risk product applications, Class I & General IVDs, the average review time is 2-4 weeks, and the maximum processing time can reach 90 days.
If it is a product that needs Approval Body audit, it needs to be determined according to the specific audit time of different AB.
A: A separate QMS certificate is required for medical devices to be registered in the UK.
A: Source of fees: MHRA
https://www.gov.uk/government/publications/mhra-fees/current-mhra-fees).
A: Yes, the MHRA allows the manufacturer to change to a new authorized representative.
A: No, a product is only allowed to have one authorized representative.
A: If the medical device product changes conform to the scope permitted by MHRA, an authorized representative can submit a change application, and after MHRA approval, it can be put into the UK market.
A: No, batch upload cannot be used for all GMDN Code combinations. The Batch Upload function is only available for a device model with a certain GMDN Code. For example, to register a GMDN Code that includes 50 different device models, you can register these 50 device models using a batch pass.
A: It is currently in a transitional phase, and if the extension regulations remain unchanged, the EU MDD/IVDD or EU MDR/IVDR device labeling and packaging in the UK will not need to be changed until June 30, 2023. As a result, the current logo and packaging (including the CE marking) will remain valid, while the name and address of the UK RP will be mandatory on the logo and packaging until 30 June 2023.










































