
Regulatory Agency website link: https://eur-lex.europa.eu/eli/reg/2017/745/oj/eng
Database link:https://ec.europa.eu/tools/eudamed/#/screen/search-device
The key EU regulatory acts governing medical devices and their implementation dates are as follows:
1. Medical Devices Regulation (EU) 2017/745, applicable from 26 May 2021;
2. In Vitro Diagnostic Medical Devices Regulation (EU) 2017/746, applicable from 26 May 2022;
3. Regulation (EU) 2023/607 of the European Parliament and of the Council of 15 March 2023, which amends the transitional provisions of the above two Regulations and extends the transition period for the Medical Devices Regulation (MDR) in phases.
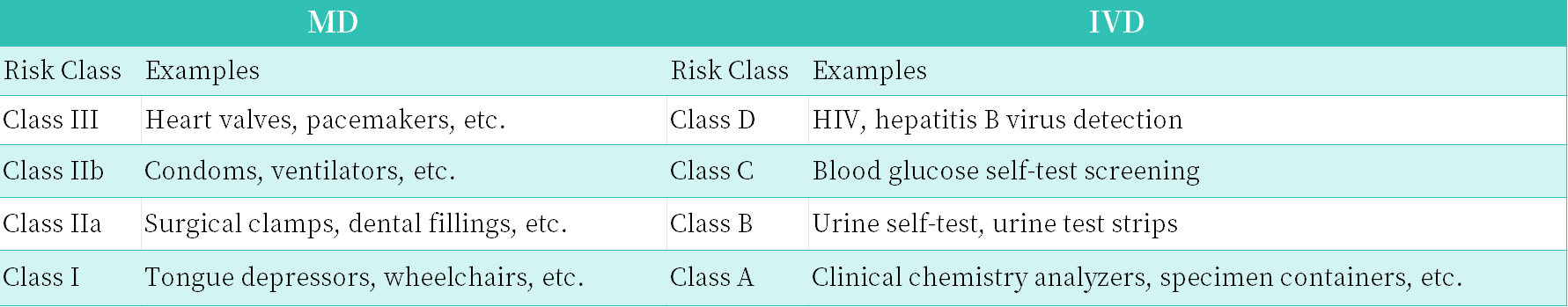
The European Union classifies medical devices and in vitro diagnostic (IVD) devices into risk categories, ranging from low to high:
Medical device classification: Class I (including general Class I, sterilised Class Is, measuring Class Im, and reusable Class Ir), Class IIa, Class IIb and Class III, where Class I represents low risk and Class III represents high risk;
IVD classification: Class A, Class B, Class C and Class D.
1. Possession of an ISO 13485 system certificate and annual audit report;
2. Appointment of a designated European Authorised Representative;
3. All medical devices must be registered in the EUDAMED database.
English.
A: Not necessarily, according to the product characteristics and products has been approved by the market, most of the devices can be selected to meet the conditions of the substantial equivalent device for comparison.
A: There is no mandatory requirement, if there is no racial difference in the device, some notified bodies also accept domestic clinical reports in China, but the trial must meet the requirements of ISO 14155:2020 and other relevant guidelines or regulations. In vitro diagnostic reagents must meet the requirements of ISO 20916:2019 in addition to ISO 14155:2020.
Wiselink has clinical trial sites in Greece, Italy, Germany and Slovenia to carry out clinical trial projects for you.
A: Labels and instructions translated in the local official language are required, and are also required to be filed with the notified body in advance.










































