The registration system of the Saudi SFDA has its unique rules, and many enterprises find it confusing when first encountering it. This article systematically sorts out all the core points of Saudi medical device registration to help you understand the access logic of the largest market in the Middle East in one go.
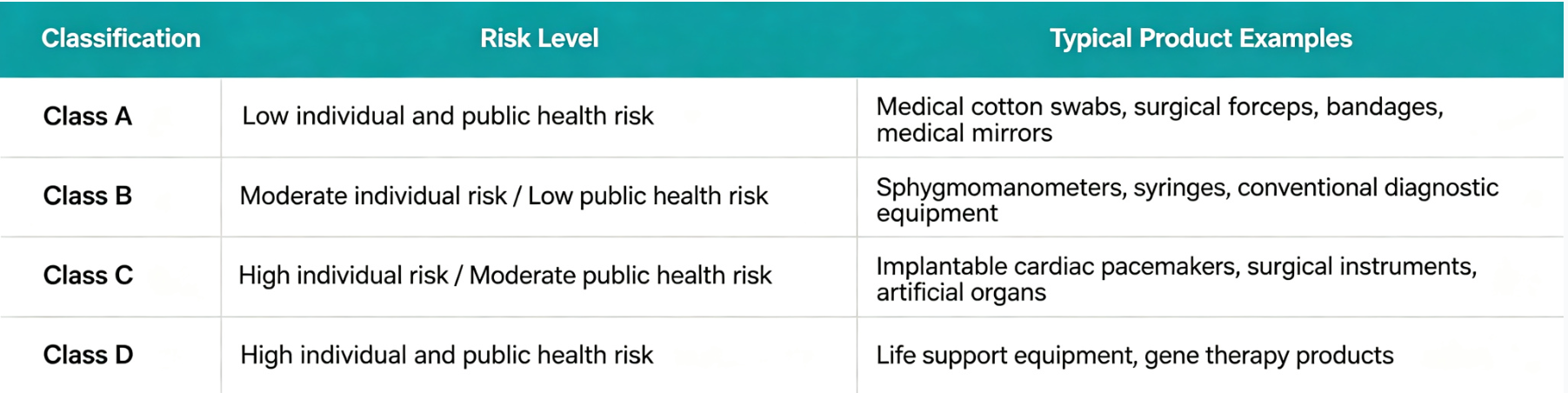
The four-level classification also applies to IVD products, such as virus detection kits, specific tumor marker detection kits, and HIV detection kits, which correspond to different categories according to their risk levels.
Core regulatory basis: Medical Devices Law + Implementing Regulation of Medical Devices Law
Official review period: approximately 35 working days (about 90 days)
Actual full-process period: approximately 7 months (including preliminary document preparation, AR authorization application, etc.)
Complete device description: including materials, human contact parts, and working principle
Design and manufacturing process description
Risk assessment report (strictly in accordance with ISO 14971)
Clinical evaluation data: if the equivalent product route is adopted, proof of technical, biological, and clinical equivalence must be provided
Essential attachment: Essential Principles of Safety and Performance Checklist, itemizing each regulatory requirement met and the verification methodLanguage requirements: Labels and instructions for household products must be bilingual (Arabic + English). Electronic instructions for use (e-IFU) require additional risk assessment (e.g., anti-tampering measures, network outage contingency plans).
Environmental adaptation: Saudi Arabia has a high-temperature and high-humidity climate. Device design must consider local environmental conditions, and transportation and storage conditions must be clearly specified in the technical documentation (e.g., temperature range of -20°C to 50°C).
Validity period: 3 years
Renewal window: Renewal can be applied for within 3 months before expiration
Renewal materials: MDMA renewal application form (submitted online via the GHAD system), copy of the original registration certificate, valid Saudi AR authorization letter (signed and stamped), manufacturer authorization letter (authorizing AR to handle renewal), and renewal fee payment receipt.As the largest medical device market in Latin America with the most sophisticated regulatory system, Brazil is the core position for domestic medical device enterprises to expand into the South American market. In celebration of Wiselink's 6th anniversary, we are launching a limited-time offer: from June 18 to July 18, consulting service fees for all risk classes of medical device products in the Brazilian market are uniformly discounted by 20%! New and existing clients are welcome to inquire!













































